自分の患者でもADPKDの人は多いが、遺伝性疾患であるので患者のケアももちろんであるが家族のケアも非常に重要な疾患である。
下図のように進行性の病気であることもわすれてはならない。

まず、今回はADPKDに関してお話しするが、ARPKD(常染色体劣性多発性嚢胞腎)もしっかりと認識しておくことは重要である。
常染色体優先遺伝の場合は下記。
常染色体上に存在する一対の遺伝子の一方に異常があれば発症する。
なので、やはり家族へのケアは注意する必要性がある。
 |
| 大塚製薬サイトより |
原因:遺伝子異常が原因
ADPKD:PKD1遺伝子とPKD2遺伝子が主役になる。このPKD遺伝子は腎臓ならば尿細管の液体のながれを感知し、肝臓ならば胆細管の胆汁の流れを感知する。流れを感知して、順序よく並んで流れをスムーズにいくように働きかけている。
※PKD1から作成されるものがPC1(polycystin1)、PKD2から生成されるものはPC2である。
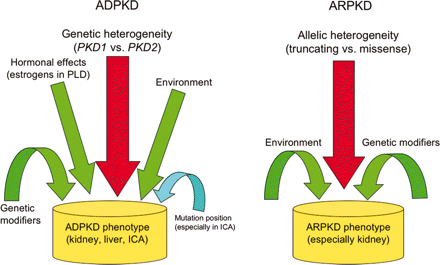
※PKDの違いによるものは下記に示す。
 |
| 杏林大学Homepageより |
 |
| 杏林大学Homepageより |

その際に治療で重要になるのが、cAMPである。
cAMPは抗利尿ホルモン(バソプレッシン)の作用を細胞内に伝える働きがある。
正常細胞ではcAMPは細胞増殖を抑制するが、ADPKDでは嚢胞細胞の数を増やしてしまう。
★また、ADPKDの発生に関しては遺伝子異常はもちろんであるが、下図に示すように多彩な原因で生じてくる。その一つが肥満である(図)。(JASN 2007)

症状に関してはほとんど無症状の場合が多い。
血尿が30%程度におこり、嚢胞感染や嚢胞出欠などがある際に生じる背部痛を契機に気づかれる症例もいる。
検査:下記が診断基準となっている。

つまり、検査ではCT検査やMRI検査が重要となる。
ここで、除外すべき疾患として記載されているように、それらの疾患の可能性はどうなのかを常に考えることは重要である。
家族内発生をしっかりと認識する事は一番重要である。
また、併存症の管理が大事である。
致死的になるものは脳動脈瘤破裂であり、だいたい10%程度の人に見られるものなので、ここに関しては把握しておく必要がある。
 |
| 杏林大学Homepageより |
2部構成に今回はしようと思う。
今回の点で大事なのは、遺伝子異常はもちろんのことADPKDを悪化させるものはないか?家族は大丈夫か?である。
